

Computer-aided or computational drug design is fast gaining prominence in many drug discovery campaigns. This approach leverages on chemical and biological information about ligands and/or biological targets to design potential drug candidates whereby the biological and physical properties of the target are studied and a prediction is made of the sorts of chemicals that might ût in to an active site. This streamlines the drug discovery and development, avoiding a large population of inactive compounds to save money, time and resources.
Today, there are many drugs which owe their discovery to the application of computational method of drug design. One of such is Dorzolamide, a carbonic anhydrase inhibitor used for the management of glaucoma. It was the first drug which resulted from structure base computational drug design approach introduced in 1995 by Merck.
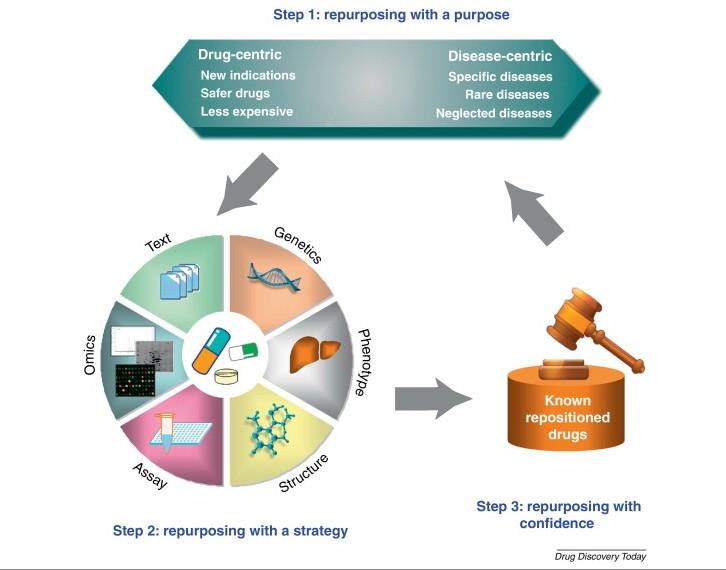
 Computer-aided drug design as a tool in drug development is being applied in the repositioning of known drugs for a different therapeutic use. Computational drug repositioning involves repurposing a known drug for new therapeutic indication, using computer technology. In other words, it involves finding new therapeutic use for already existing drugs.
Computer-aided drug design as a tool in drug development is being applied in the repositioning of known drugs for a different therapeutic use. Computational drug repositioning involves repurposing a known drug for new therapeutic indication, using computer technology. In other words, it involves finding new therapeutic use for already existing drugs.
Examples of repositioned drugs
There are many examples of drugs successfully repositioned. A typical example is the phosphodiesterase inhibitor, Sildenafil, sold as Viagra, which was initially developed for treating pulmonary arterial hypertension (angina) but later repositioned for the treatment of erectile dysfunction. Another well-known example is Thalidomide developed by the German drug company, Chemie Grunenthal in 1957. It was primarily prescribed as a sedative or hypnotic. Afterwards, it was used against nausea and to alleviate morning sickness in pregnant women. Due to its teratogenic effects as seen in the malformation of limbs in infants, it was withdrawn from the market in 1961 and later repositioned for the treatment of certain cancers (multiple myeloma) and in complication of leprosy.
It is worthy of note that drug repositioning may be achieved with or without computational design tools. In the past, some drugs formerly indicated for the treatment of certain ailments or used for entirely different purposes were discovered serendipitously or deliberately repositioned to be useful for other therapeutic indications. An example is the sulphur mustard which was used as a biological weapon during the second world war, but later repositioned without the use of computer-assisted drug modelling, serving as the template for the development of the nitrogen mustards (DNA alkylating agents) such as Melphalan used in cancer chemotherapy.
For a drug to be successfully repositioned, adequate information on the biological activity of the drug and/or target and its involvement in the pathology of the disease condition must be known. The process of gathering such functional information of the biological target is called “target validation” in pharmaceutical industry parlance.
The application of computational drug repositioning as an alternative approach in drug development has been made possible by increase identification of molecular targets, elucidation of the 3D structures by X- ray crystallography and Nuclear magnetic resonance (NMR), availability of commercial, private or public databases (for biological targets and ligands), and availability of computer-aided drug design software.
Approaches to computer-aided drug design
Depending on the availability of structural information, a structure-based approach or a ligand-based approach is used. Structure-based computer aided drug design depends on the information of the target protein structure obtained from X-ray crystallography, NMR or homology modelling to calculate interaction energies for all tested compounds. This approach involves “docking” a process of ligand binding to its receptor or target protein, to identify and optimize drug candidates by examining how the drug interacts with its target and modelling molecular interactions between ligands and target macromolecules.
According to Anson et al (2009) the conventional methods (High Throughput screening) of drug discovery is a lengthy, “expensive, diûcult, and ineûcient process” with low rate of new therapeutic discovery. The average time to develop a new drug has also increased over time (Hurle, Yang et al. 2013). In 2010, it was estimated that the cost of research and development of new molecular entities (NME) was US$1.8 billion (Paul et al 2010). Very few compounds, out of hundreds of thousands tested in animals, reach human clinical trials. This represents an enormous investment in terms of time and money and other resources.
For a very long time, there have been many propositions in improving the efficiency in the drug discovery and development process in the pharmaceutical industry. A Pharma report by PriceWaterhouse coopers in 2005 : “An Industrial Revolution in R&D” stressed the reality that pharmaceutical industry needs to find means of improving efficiency and effectiveness of drug discovery and development in order to sustain itself.”
The report emphasized growth and value of computational approaches to address this issue and projected that it will become dominant approach of drug discovery in the nearest future.
Benefits of computational drug repositioning
There are many benefits presented by computational drug repositioning compared to conventional methods of drug discovery, which involves a trial-and-error approach of lead identification from natural sources or high throughput screening (HTS) of large chemical libraries in vitro.
Firstly, in computational drug repositioning, the pharmacological profile (Pharmacokinetics, safety profile, toxicology and drug interactions) of the drug to be repositioned is well-known (Lu, Agarwal et al. 2012).
Secondly, it is both cost effective and time-efficient as it increases the effectiveness and efficiency of drug discovery at a lower price and decreases the use of animals in the process of lead identification and optimisation (Kapetanovic 2008). The estimated time required for repositioning of a known drug for a new clinical indication can be as low as three years (Hurle, Yang et al. 2013).
References
Anson D, MaJ, HeJ-Q (2009). “Identifying Cardio-toxic Compounds”. Genetic Engineering & Biotechnology News. Tech Note 29 (9) (Mary Ann Liebert). pp. 34– 35. ISSN1935-472X.OCLC77706455.
Hurle, M., L. Yang, et al. (2013). “Computational Drug Repositioning: From Data to Therapeutics.” Clinical Pharmacology & Therapeutics 93(4): 335-341.
Kapetanovic, I. (2008). “Computer-Aided Drug Discovery and Development (Caddd): In Silico-Chemico-Biological Approach.” Chemico-biological interactions 171(2): 165-176.
Paul S, Mytelka D, Dunwiddie C, Persinger C, Munos B, Lindborg R, Schacht A. (2010). “How to improve R&D productivity: the pharmaceutical industry’s grand challenge”. Nature Reviews. Drug Discovery9 (3): 203–14.
Yunusa, A. and Bello, S.O. (2015) “Computational Drug Design: An Approach In Drug Re-Positioning-A Review” International Journal of Scientific Research Engineering & Technology (IJSRET), ISSN 2278 – 0882 Volume 4, Issue 8.
Wikipedia: Drug discovery. Source: https://en.wikipedia.org/wiki/Drug_discovery?oldid=697280369










{kind=link}